Laboratory for Biomolecular Modeling

Photo by Raphaël Biscaldi on Unsplash
Our Lab
The main goal of our laboratory is to understand the physical and chemical properties of complex biological systems, in particular their function emerging from structure. To address these questions, we use and develop a broad spectrum of computational tools fully integrated with experimental data. Multiscale models and dynamic integrative modeling are used to investigate the assembly and function of molecular assemblies mimicking conditions of the cellular environment.
Research focus

Integrative structural biology
The structural determination of large molecular complexes is a formidable challenge, as they can exist in multiple functional conformations and compositions, they can interact with biological membranes, they can be regulated by post-translational modifications (PTMs) and are affected by the crowded conditions of the native environment.

Methods development
We could advance in our investigation thanks to the use and development of state-of-the-art molecular modelling and simulations at different levels of resolution and theory, which allow us to explore dynamic features that are usually non accessible to experimental techniques.

Molecular bioengineering
We aim at exploiting the fundamental knowledge acquired about structure and dynamics of biological systems to control, modulate and interfere with their properties for therapeutic intervention developing structure-based molecular entities and for biotechnological applications.

Experimental lab
We are interested in the structural characterization of macromolecular complexes with various biological functions. In order to do this, we express and purify recombinant proteins and study their 3-D structure using X-ray crystallography, NMR and cryoEM. We also characterize them biophysically using CD, MALS, MS and Mass Photometry. Complexes (either with other proteins, nucleic acids, peptides, or small molecules) are studied using EMSA, ITC, SPR and fluorescent polarization. Our experimental results are fully integrated with the computational results and tools developed in the computational part of the LBM.
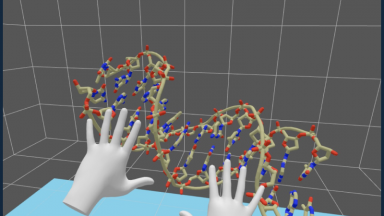
Technologies for molecular visualization
We develop methods for visualization and handling of molecules in augmented and virtual reality, advocating for web-based programming and solutions.
Contact
Head of the Laboratory: Prof. Matteo Dal Peraro
Office : AAB 0 48
Phone : +41 21 693 18 61
Administration: Magali Masson
Office: AAB 1 43
Phone: +41 21 693 2152
Mailing Address
EPFL – SV – IBI – UPDALPE
Station 19
CH-1015 Lausanne